Host Epigenetic Response to Infection:
The function that a cell performs is dictated by the sum total of the chromatin organization and the associated epigenetic modifications of each individual gene in the genome (epigenome). As the function of a cell during development and differentiation is determined by its microenvironment, any factor that can alter this microenvironment should be able to alter the epigenome of a cell.
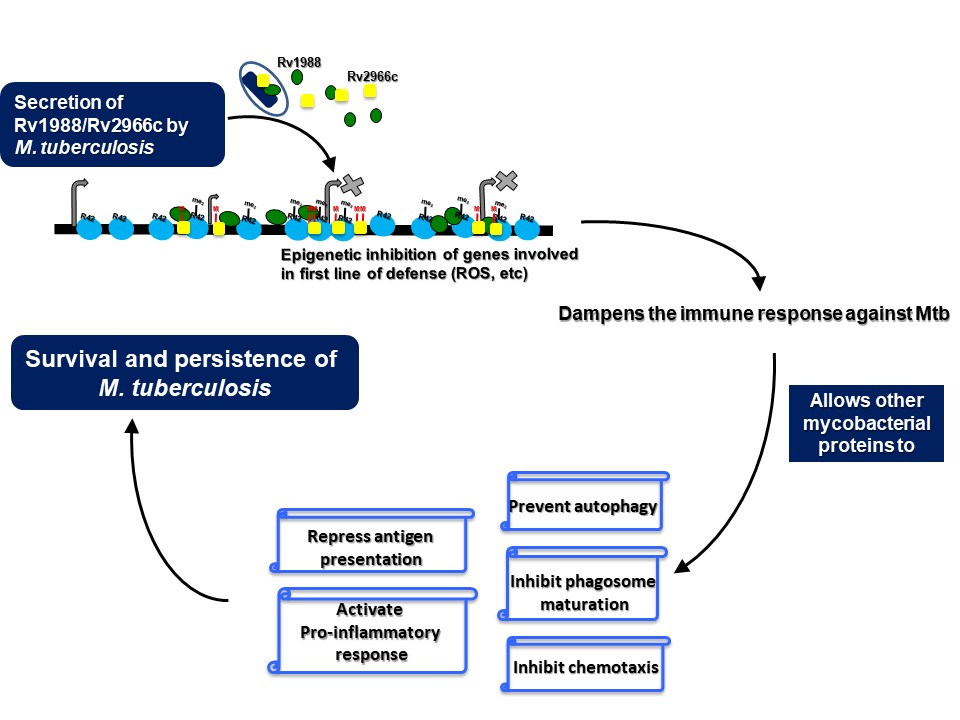
The interaction of a pathogen with its host exemplifies a tussle wherein the pathogen, like M. tuberculosis, tries to hijack the host cellular machinery and dampen the immune response for its survival whereas the host tries to eliminate or neutralise the pathogen. This interaction is not just limited to eliciting an immune response. it is also manifested through changes in specific cell-signalling cascades and change in expression of multiple genes amongst others. Changes in gene expression are possible only through alteration in the epigenetic landscape (epigenome) of a cell because epigenetic modifications like DNA methylation and histone modifications play an important role in gene regulation. Studies from our laboratory has shown that pathogens exploit the host epigenome plasticity to ensure their successful survival. We have identified DNA and histone methyltransferases from M. tuberculosis that functions to modulate the host epigenome. Rv1988 was found to be a mycobacterial methyltransferase that modulates the host epigenetic circuitry by methylating a novel, non-canonical arginine, H3R42 in histone H3. In another study, we have shown that the mycobacterial protein Rv2966c methylates cytosines present in non-CpG context within host genomic DNA upon infection. In addition, genome-wide DNA methylation changes in macrophages infected with the M. tuberculosis were identified. Hotspots of differential DNA methylation were enriched in genes involved in immune response and chromatin reorganization. Importantly, DNA methylation changes were observed predominantly for cytosines present in non-CpG dinucleotide context. All these studies indicate the use of non-canonical epigenetic mechanisms by pathogenic bacteria to hijack the host transcriptional machinery.
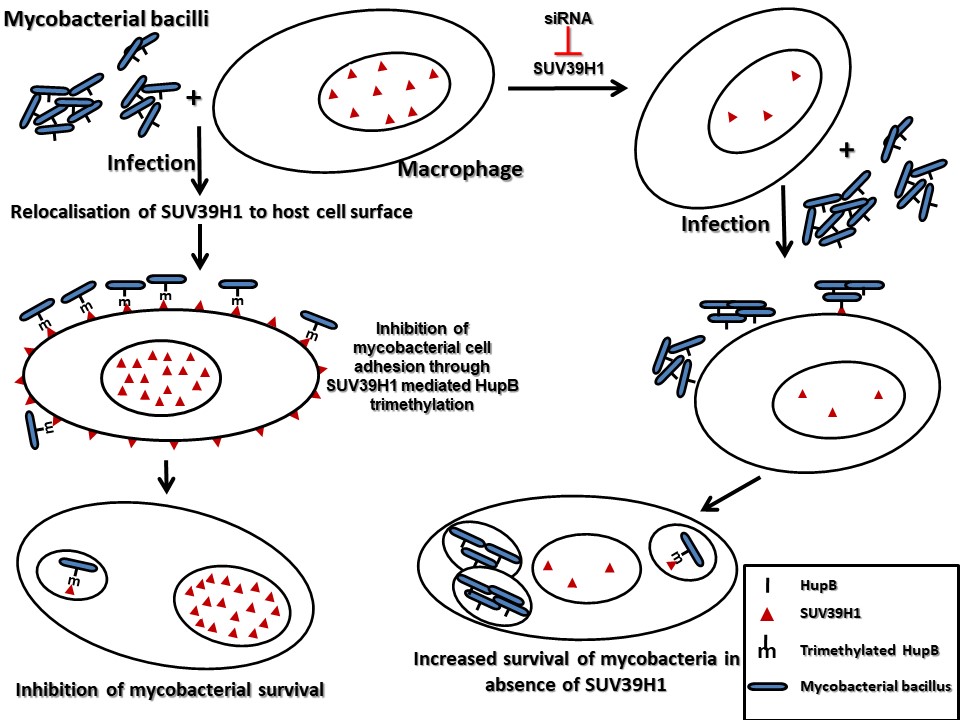
Our studies have also uncovered novel surrogate function of an epigenetic effector molecule in the host defense against pathogenic bacteria. A homolog of the Drosophila protein, Suppressor of Variegation 3(9), SUV39H1, is an evolutionary conserved protein. In all the previous studies that have examined suppressor of variegation 3(9) or its homologs, this protein has been shown to function by modulating chromatin organization, through its role as a histone H3K9 methyltransferase in the nucleus. We show that during infection, SUV39H1 is relocalised to the cell surface where it interacts with the mycobacterial bacilli and methylates the mycobacterial cell surface protein HupB, a histone like protein from mycobacteria. SUV39H1 mediated HupB methylation inhibits the adhesion capabilities of the mycobacterial bacilli and interfere with their ability to interact with the host cell. This surrogate role of SUV39H1 as a methyltransferase of the mycobacterial protein HupB, adds a novel epigenetic paradigm to the duel between the host cell and the infecting pathogen.
These studies opened new avenues to better understand host–pathogen interactions and to develop novel, more effective antimicrobial strategies.have
Chromatin organisation and genomic imprinting
The fusion of two haploid cells (a sperm and an egg) to produce the diploid zygote heralds the development of a new organism. According to Mendel's laws of inheritance the genetic information contributed by the sperm (from the father) and the egg (from the mother) is equal. This would in effect mean that given the right conditions, fusion of two sperm nuclei or two egg nuclei should lead to normal development of an organism. However, this was found not to be the case in mammals, some insects and plants. Mouse embryos with two paternal copies (androgenetic) or two maternal copies (parthenogenetic) of the genome die very early during embryonic development (Barton et al 1984 Role of paternal and maternal genomes in mouse development Nature 311(5984):374-6; Surani et al 1984 Development of reconstituted mouse eggs suggests imprinting of the genome during gametogenesis. Nature 308(5959):548-50; McGrath and Solter 1984 Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37(1):179-83). Therefore, in mammals, maternal and paternal contribution to the progeny is not equal and for normal development contribution from both parents is required. This non-equivalence of the parental genomes is epigenetic in nature and forms the basis of the phenomenon known as genomic imprinting.
For imprinted genes one copy of a DNA molecule is transcribed and the other copy is silent. What distinguishes two molecules of the same DNA sequence within the same nucleus? The answer lies not in the genetic information encoded by the DNA sequence but in its epigenetic modifications. These modifications include differential modification of the DNA itself and differential organisation of DNA within the chromatin scaffold. One important modification of DNA known in mammals is methylation of cytosine residues in the CpG dinucleotides. However, methylation does not answer all mechanistic questions related to imprinted genes. If one allele of a gene becomes methylated what prevents methylation of the other allele? Does the unmethylated allele carry another epigenetic imprint? To investigate the mechanisms of genomic imprinting, it would therefore be essential to identify the alternative imprint on the unmethylated allele.
It has been found that within the differentially methylated regions of some imprinted genes there are sub-regions that also show differential organisation of chromatin. Interestingly, the regions which show differential chromatin organisation have also been genetically defined as Imprinting Control Regions (ICRs) and shown to control the imprinting status of genes within that locus. In all the imprinted loci analysed, specialised chromatin structures were found exclusively on the unmethylated allele. This suggests that for these regions, methylation and specialised chromatin structures are mutually exclusive and reflects alternate epigenetic states. How is this mutual exclusiveness between methylation and specialised chromatin structures maintained? Do the chromatin modifications on unmethylated allele prevent it from getting methylated? More importantly, during development when are these chromatin structures established? Can such chromatin conformations behave as imprints marking the parental alleles? The aim of the ongoing project is to answer these questions and identify the role of chromatin organisation in mechanisms underlying genomic imprinting in particular and as a epigenetic regulator of gene expression in general (Feil and Khosla 1999, John et al 2001, Sowpati et al 2008, Thamban et al 2019a).
Epigenetic Inheritance
The inheritance of epigenetic modifications across multiple generations results from events during germ cell development and fertilization that allow epigenetic marks acquired during the life time of an organism to be passed on to the progeny. Epigenetic inheritance could be intergenerational as has been documented for genomic imprinting or transgenerational (see figure on transgenerational epigenetic inheritance) where epigenetic modification acquired in one generation is inherited unchanged across several generations. Work from our laboratory has shown the role of DNMT3L in epigenetic inheritance.
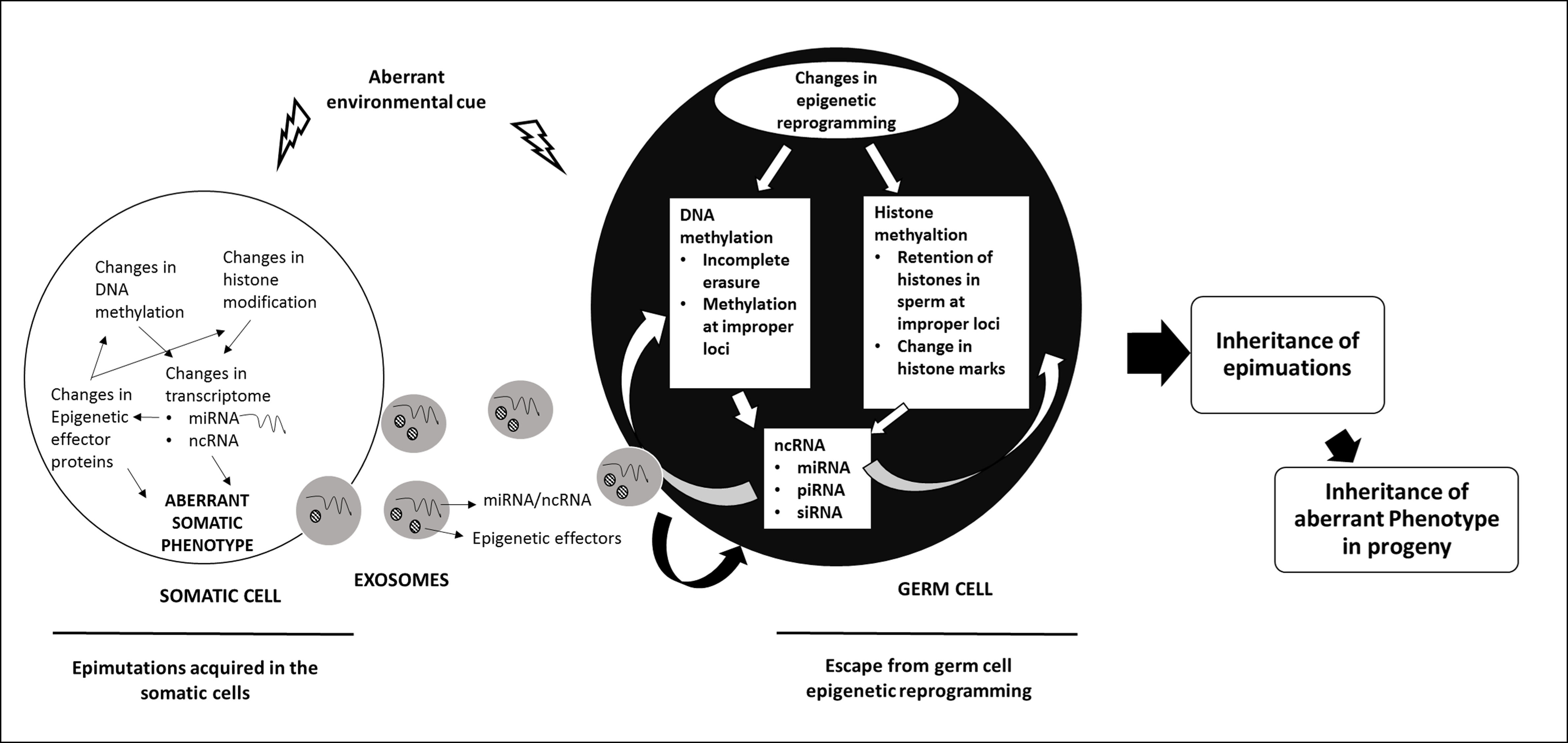
Figure Legend: Mechanisms of Transgenerational Inheritance. A hypothetical model describing the possible mechanisms by which epimutations acquired by the somatic cells are transmitted to the germ cells and inherited to the next generation (ref: Thamban et al 2019b)
DNMT3L is an epigenetic modifier that interacts with de novo methyltransferases DNMT3A/3B and histone H3. Work from our laboratory has indicated a role of DNMT3L in nuclear reprogramming. Nuclear reprogramming observed in both DNMT3L expressing Drosophila and mammalian cells overexpressing DNMT3L was gradual and progressive. In Drosophila, ectopic DNMT3L expression caused progressive misregulation of genes and by 5th generation a very large number of genes were aberrantly expressed. Some of the transgenic flies expressing DNMT3L developed melanotic tumors and did not survive. The progressive misregulation of genes across several generations in transgenic Drosophila involved germline passage through meiotic divisions. Most epigenetic marks are normally erased and reset in the germline. Therefore, any epimutation acquired during one generation would be lost in the germ cells and hence not inherited. Even for imprinted genes, the epigenetic imprints inherited from the previous generation are erased before new epigenetic modifications determined by the sex of the individual are initiated. However, we observed a progressive decrease in the genome-wide levels of H3K4me3 and H3K36me3 across more than one generation. This accumulated decrease in these modifications (or accumulation of epimutations) suggested that in the presence of DNMT3L new epigenetic modifications were being added over and above of what was inherited from the previous generation. It is possible that epimutation accumulation could be incapacitation of protein(s) responsible for erasure or resetting of epigenetic modification, as a secondary event in Drosophila expressing DNMT3L. Hence, in each generation DNMT3L would keep adding new epimutations to the ones inherited from the previous generation. On the other hand, it is possible that the presence of DNMT3L prevented erasure of epimutations inherited from the previous generation. In this scenario, epigenetic modifiers responsible for setting up of new epigenetic marks in the germ cells would add on to the epigenetic modifications that were not erased due to the presence of DNMT3L. Further work is being carried out to examine these hypotheses (Basu et al Scientific Reports 2016, Thamban et al 2019b)
|